NMR-assisted MD and MREMD simulations with time-averaged restraints
Note! Simulations of this type are available in the Advanced mode only. This tutorial covers MREMD simulations; MD simulation settings are part of it.
First, select the Advanced mode and then MREMD or MD (MREMD has been selected in the example below).

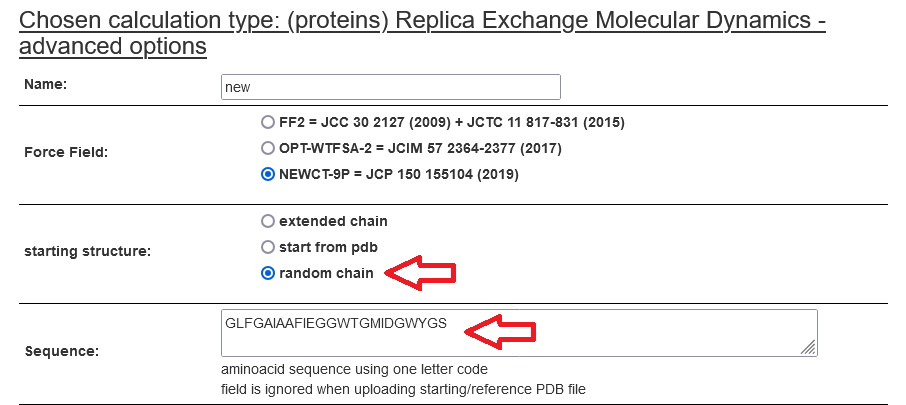
Subsequently, define the sequence, starting conformation, and reference structure if required, as shown below.
Mark Use NMR restraints, specify the restraint format and restraint file. Optionally, restraint-function parameters can be changed from the default values.

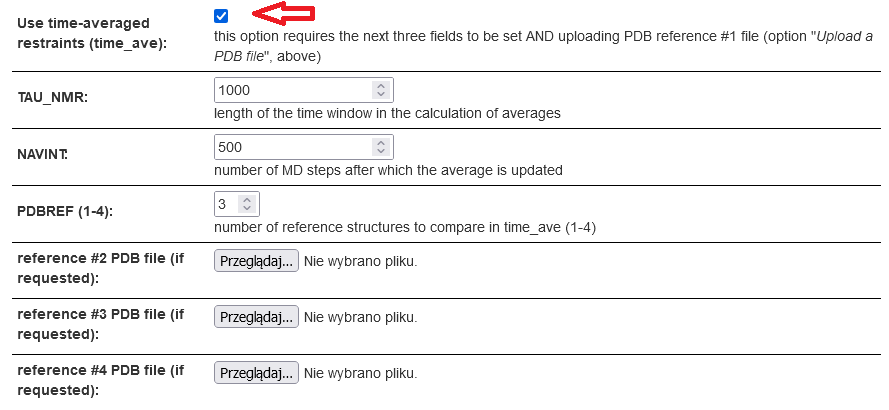
Mark Use time-averaged restraint. The length of memory window (TAU_NMR) and number of divisions of the memory window (NAVINT) into segments after which global average update is performed can be changed from the default values. Additionally, more reference structure (from #2 on) can be selected and the respective PDB files uploaded; this choice is particularly useful if a multistate protein is studied. Having completed the settings, click Save & Submit

The Replica Exchange options will appear, which enable us to change the temperatures and number of replicas per temperature from the default values. However, please note that the default values have been well optimized and tested. Having optionally changed the default values click Save and Submit again.
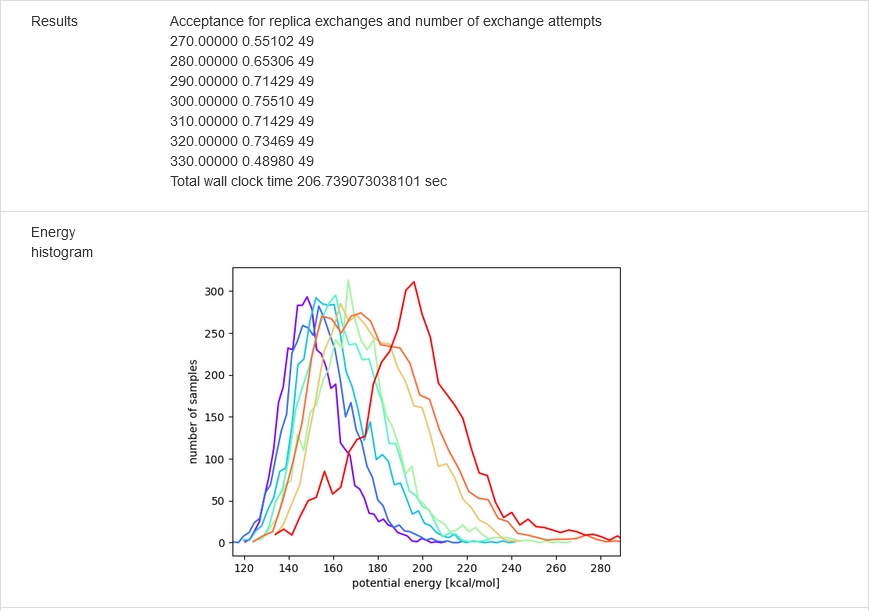
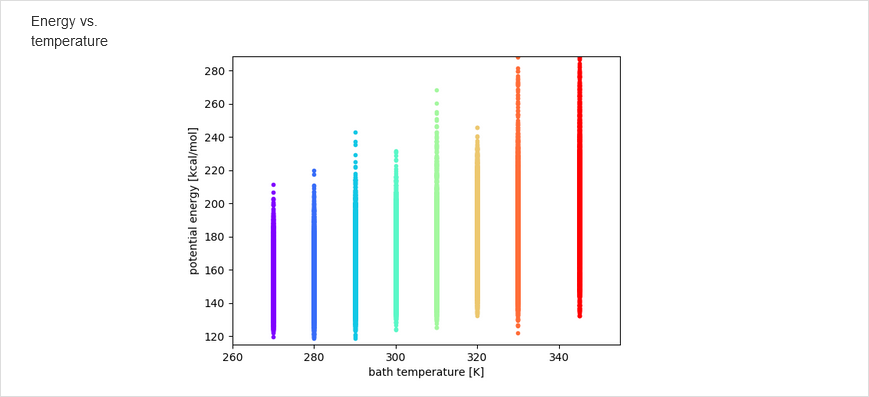
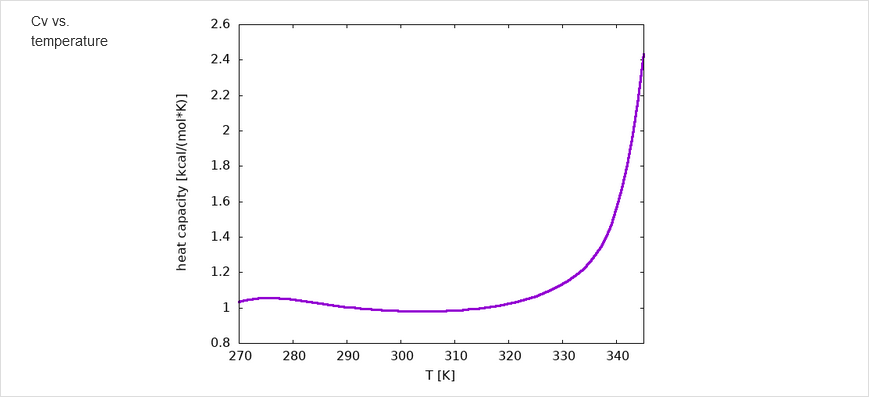
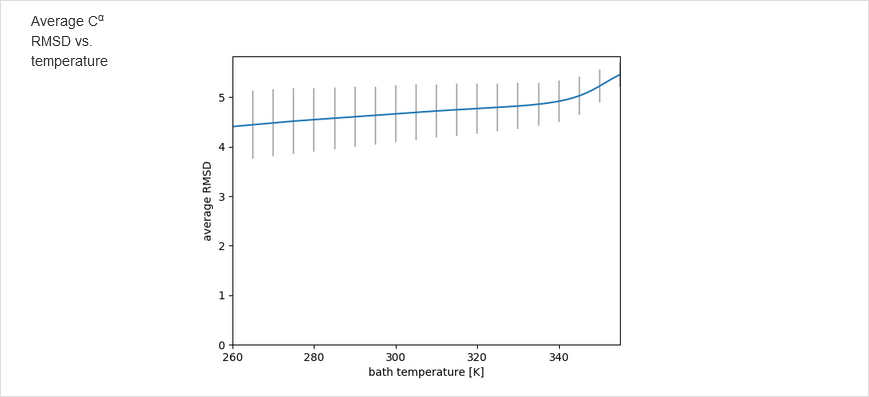
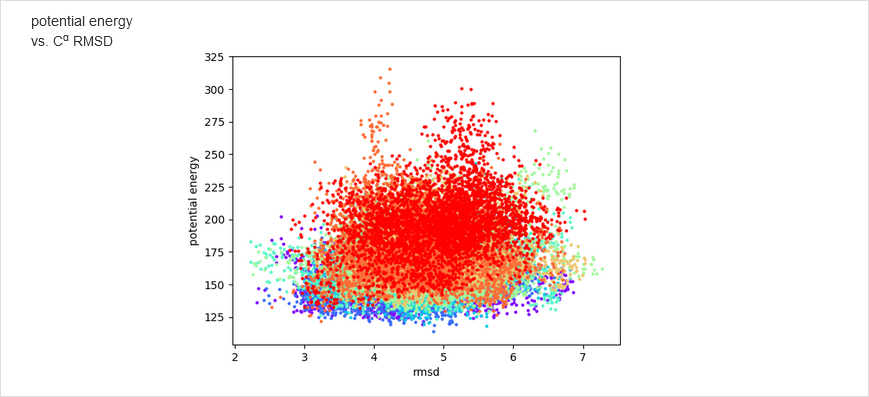
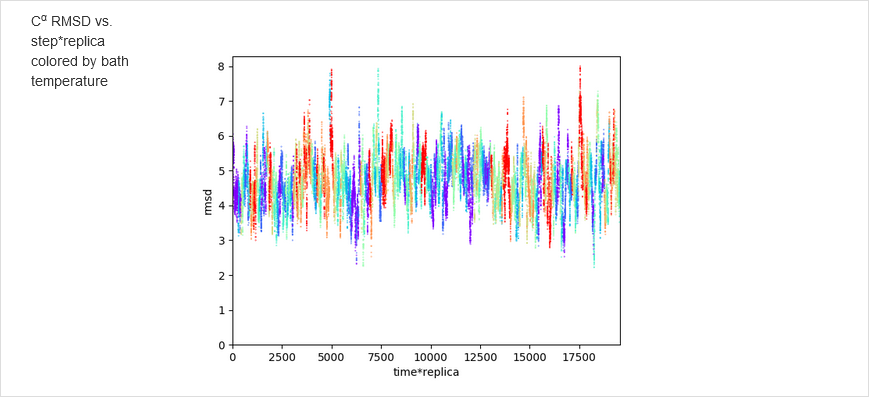
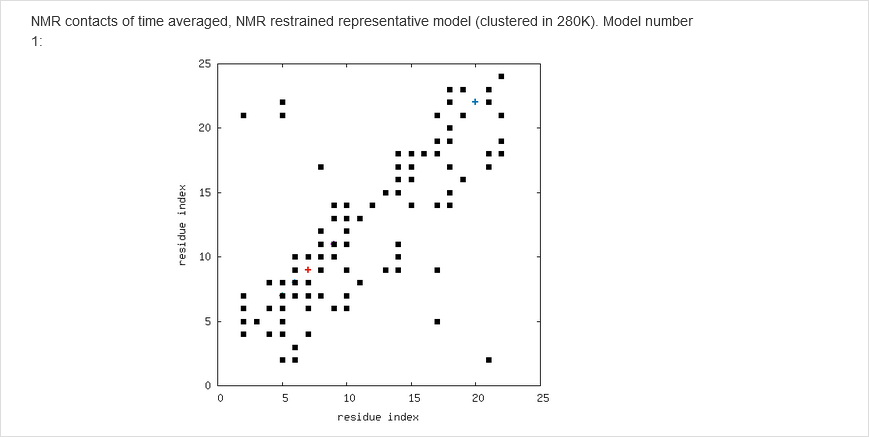
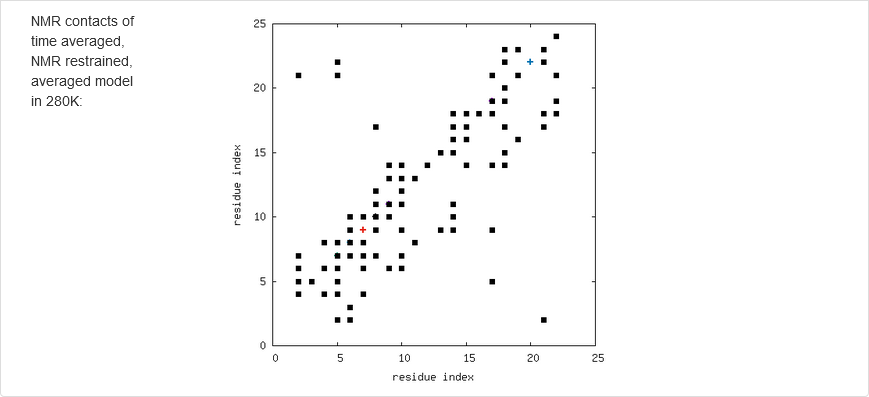
The results of calculations will appear in the graphical form, as shown below, the figure legends being self-explanatory. For some browsers it may be neccessary to refresh the webpage to display previously shown model/reference structure (the broweser console displays a warning: Too many active WebGL contexts. Oldest context will be lost.)