PEPTIDE AGGREGATION SIMULATION
In this tutorial, we will describe how to run MREMD simulations for peptide aggregation.Only the sequence of a single peptide chain is required as input. The UNRES server will generate and distribute the peptide molecules in the simulation box automatically. The other simulation parameters can be changed from the default values if required.
First, choose the AGGREGATION type simulation:
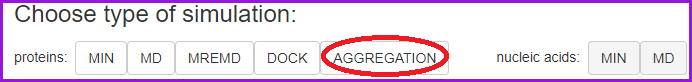
In the Basic mode, input the peptide sequence and concentration (default 0.002 mol/dm3).
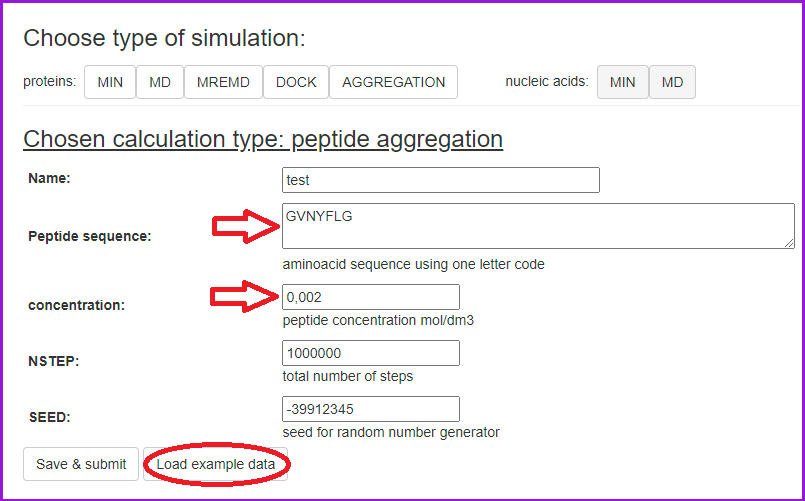
Start the simulation by clicking Save & Submit:

In the Basic mode, 8 peptide molecules are placed in the simulation box. To change this, the Advanced mode is required.
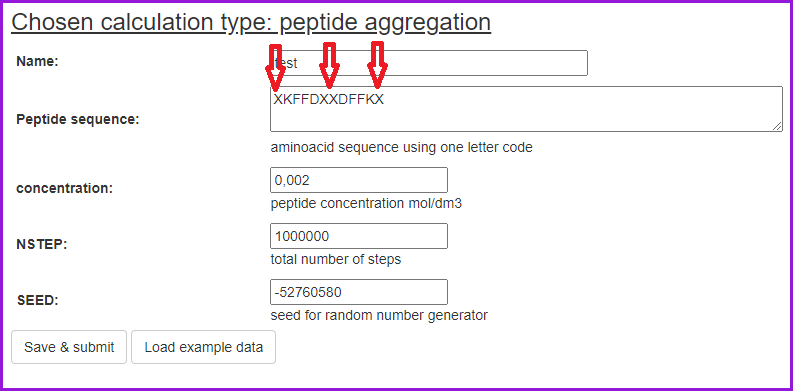
Peptide mixture can also be generated by inputting the extended sequence, in which individual peptide molecules are separated with "XX", as shown in the example below. Note that the concentration will then pertain to the total concentration of ALL peptide molecules and the ratios will be determined by the extended sequence. For example, if two peptides (A and B) are in solution at the 2:1 ratio, input the sequence as X(A-sequence)XX(A-sequence)XX(B-sequence). The concentrations of A and B will then be 2C and C, respectively, where C is the total peptide concentration.
The simulations can also be run in the Advanced mode.

Here, more options are available, including defining the number of molecules:
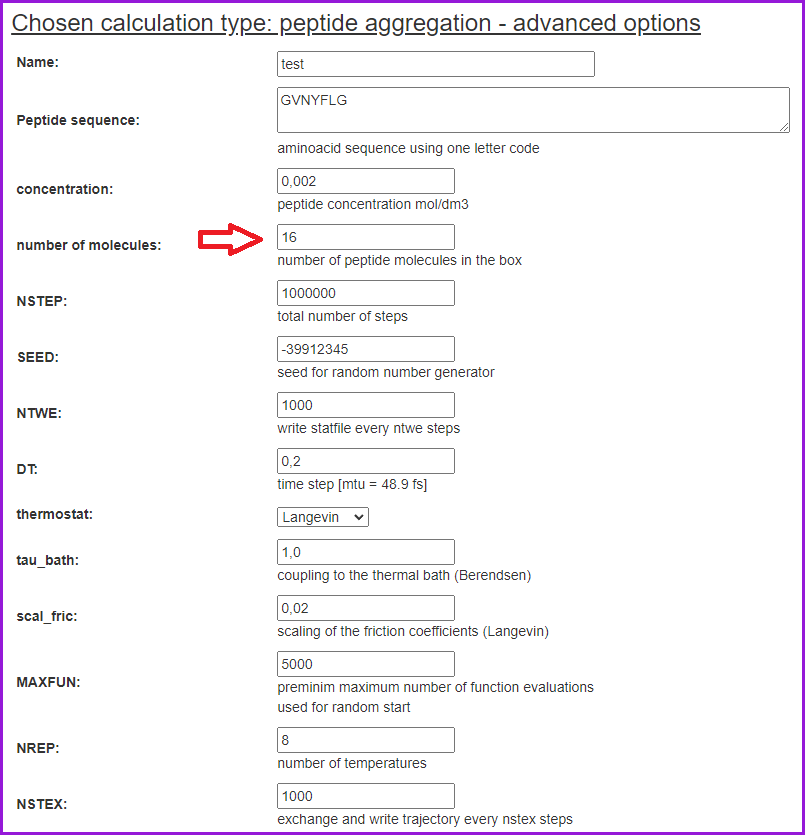
After setting all the options, we can click on the Save & Submit button. The Replica Exchange options will appear, which enable us to change the temperatures and number of replicas per temperature from the default values. However, please note that the default values have been well optimized and tested. Having optionally changed the default values click Save and Submit again.

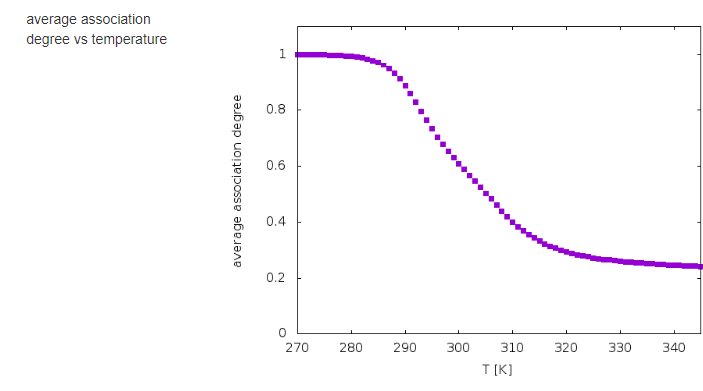
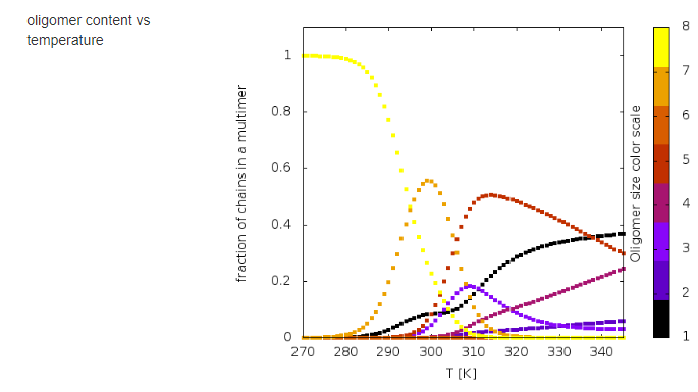
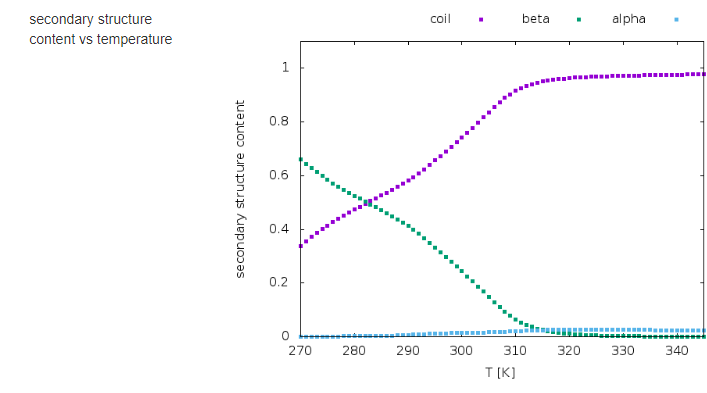
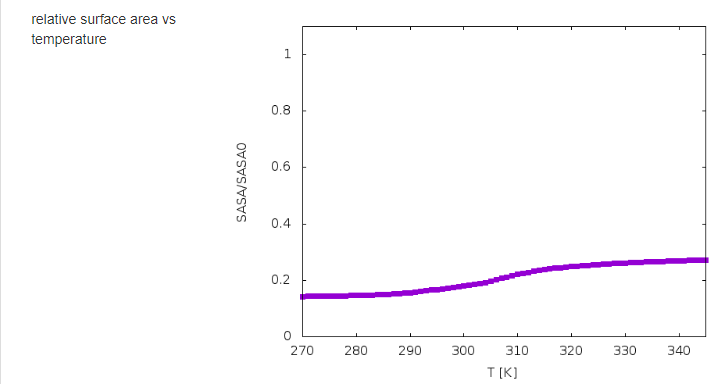
The results of calculations will appear in the graphical form, as shown below, the figure legends being self-explanatory.